Authors:
Tyler Ngo tdngo@ucsd.edu
Ethan Lee e3lee@ucsd.edu
John Driscoll jjdrisco.edu
Mentor: Tiffany Amariuta tamariutabartell@ucsd.edu
Abstract
We explore the connections between common genetic diseases and gene expression, specifically Alzheimer’s and age-related diseases to piece together information that improves genetic understandings of disease mechanisms. To accomplish this, we use colocalization and gene set enrichment analysis to understand relationships from a high level and uncover highly enriched pathways in gene subsets. In our deep dive into large genetic datasets, we uncover snippets of how different diseases are linked and find genetic evidence for disease mechanisms discovered environmentally. We do this by comparing SNP significance in narrow windows across Alzheimer’s and related diseases, against other diseases unrelated to aging. We hope that finding evidence in this manner will reinforce the significance of colocalization and gene set enrichment analysis as indispensable tools for genetic discovery.
Introduction
Understanding more about aging-related diseases, such as Alzheimer’s or dementia, is a crucial stepping stone for medical advances and increased prevention. In this study, we piece together underlying genetic factors that define them.
We use various methods to understand how diseases are connected at the genetic level. The central method in this process, colocalization analysis, is a powerful tool that will allow us to see if numerous traits share any causal genes. Additionally, we employ principal component analysis (PCA) to visualize patterns in our genetic datasets. PCA allows us to reduce the dimensionality of complex data matrices and identify clusters, providing valuable insight into the genetic relationships among the targeted diseases.
We also use gene set enrichment analysis (GSEA) to identify genes within gene sets that have a stronger effect subset by our phenotype data, in this case, by gender data publicly available for the 1000G dataset. We then cross-reference our significant gene results from our colocalization and the GSEA to make notable conclusions.
By implementing these analytical approaches, our study aims to establish a connection between the genetic basis of Alzheimer’s and age-related conditions. By figuring out how diseases are related genetically and finding shared pathways, researchers can determine better treatments for them. Discoveries made in this way can significantly change how age-related diseases are diagnosed, prevented, and treated.
Methods
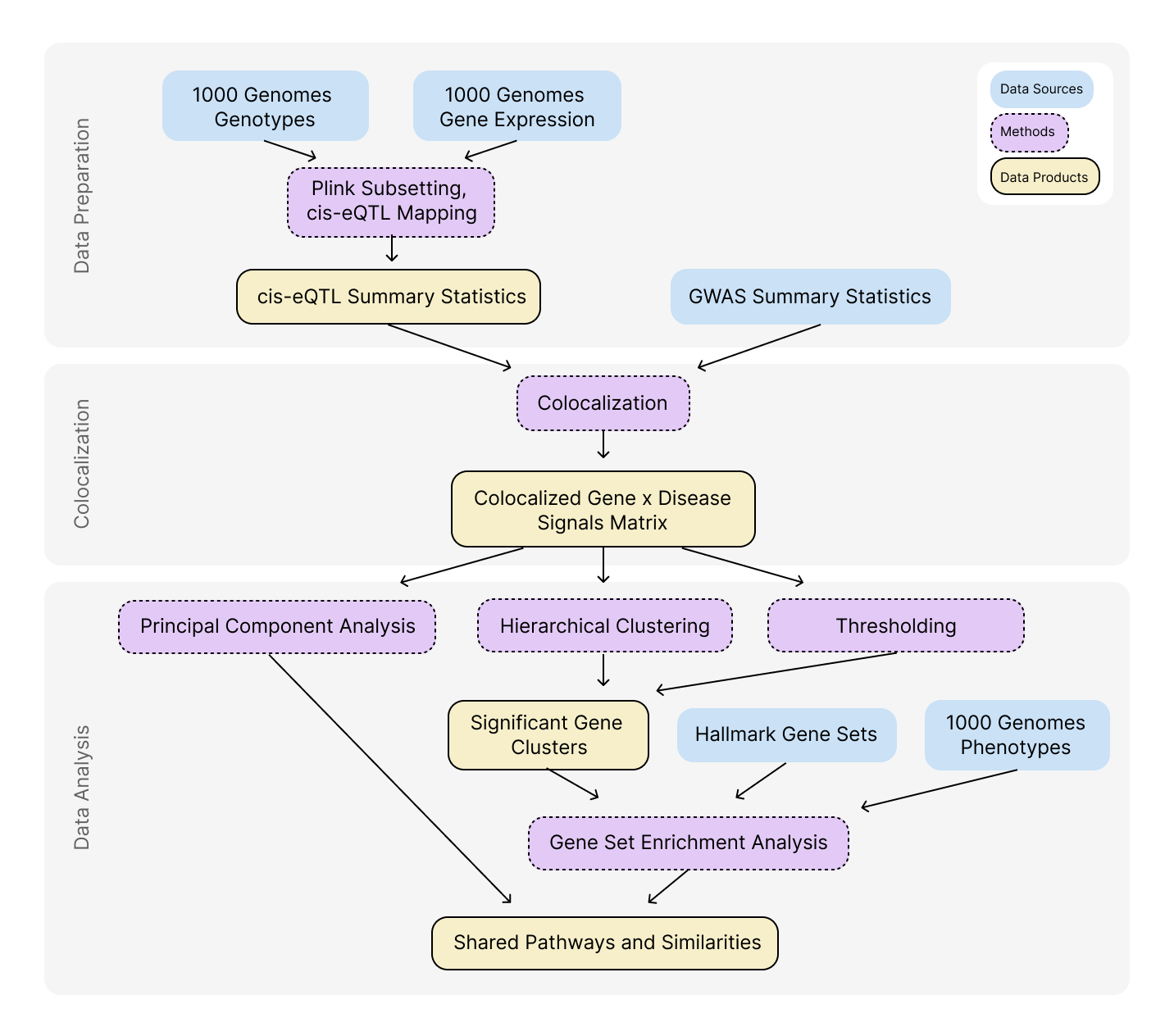
Data Collection and Preprocessing
To investigate our main interest in this work, aging-related disease, we pull publically available GWAS data, genotype data, gene expression data, and gene set databases. We find relevant GWAS summary statistics for our diseases of interest from the GWAS Catalogue genotype and gene expression data from the 1000 Genomes dataset, and gene sets from the Molecular Signatures Database (MSigDB). To use these data sources in our analysis, we had to format, label, normalize, and clean up the datasets using various techniques in Python.
Colocalization Analysis
We conduct colocalization analysis (COLOC) by superimposing GWAS summary statistics for several aging-related diseases and against cis-eQTL summary statistics generated using gene expression and genotype data from the 1000 Genomes dataset. Colocalizing the summary statistics allows us to assess the statistical likelihood of there being a shared causal variant between two independent associations: disease status and the expression of a particular gene. We implemented the analysis using the "coloc" package in R, and ran the analysis in batches due to the large size of the GWAS data. The resulting matrix contains the results of statistical tests, which identify the strength of evidence for a shared causal variant at points of interest in the genotype data.
Principal Component Analysis and Hierarchical Clustering
One approach to extracting results from our COLOC matrix was Principal Component Analysis (PCA). PCA serves as a dimensionality reduction technique, which allows for the analysis of potential patterns or relationships within the genotype architecture behind the aging-related diseases of interest. We apply PCA through code configured in Python and find that the first component accounts for 60% of the variation in the matrix, effectively separating two aging-related diseases from the rest, allowing us to subset and study their similarities with various analysis and visualization techniques. Additionally, we employ hierarchical clustering to analyze patterns within the results data further. This allowed us to isolate areas of shared high causal variant likelihood, which may indicate that the same signal is shared across diseases.
Gene Set Enrichment Analysis
The final crucial method of our project is gene set enrichment analysis (GSEA), which we perform on subsets of genes expressed in individuals from the 1000 Genomes dataset based on the Molecular Signatures Database (MSigDB) Hallmark gene set. GSEA identifies biological pathways or gene sets significantly enriched in a gene expression dataset, meaning they likely play a role in disease mechanisms. We generated enrichment scores among other values for each gene set and individual genes for subsets generated by PCA and hierarchical clustering. The results of GSEA allow us to understand which biological functions are implicated by the colocalization analysis, giving us an understanding of the pathways activated behind aging-related diseases.
Results
Colocalization Matrix
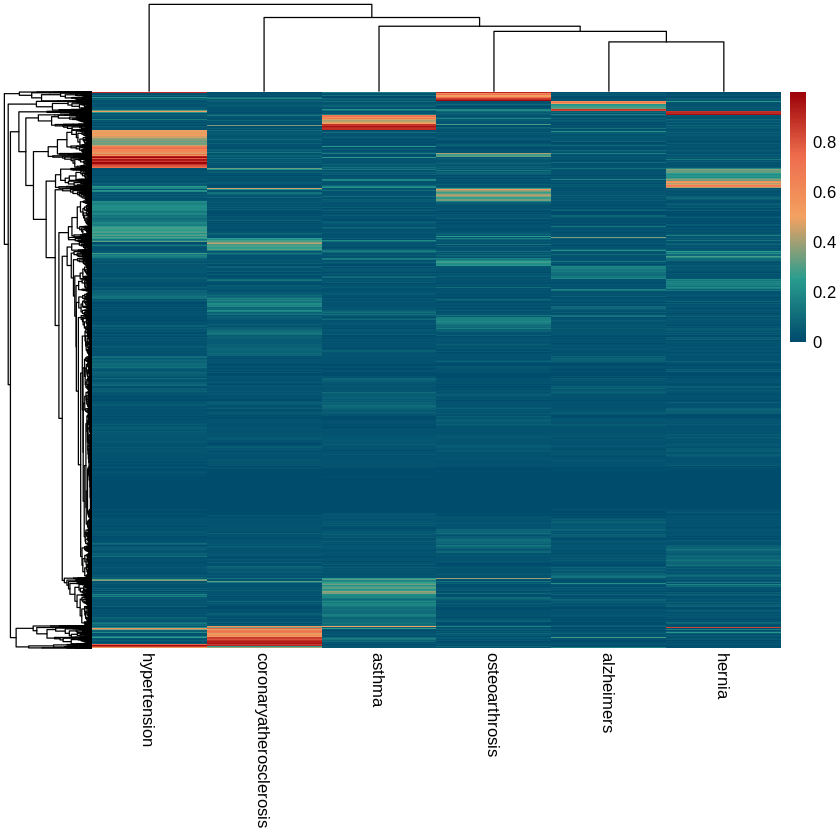
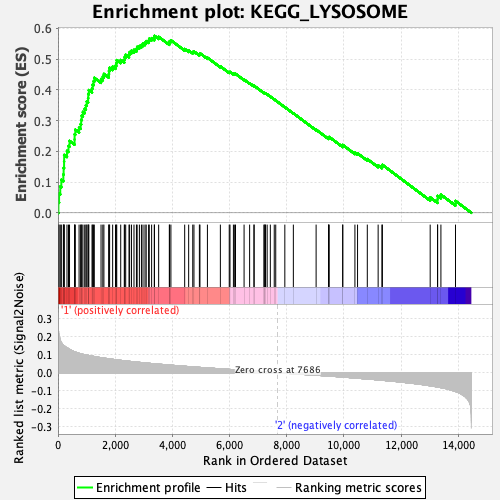
Our colocalization output contained prior probability values that ranged from 0 to 1 with the large majority falling between 0 and 0.2, indicating that evidence of shared causal variants is weak for a majority of the gene windows. When we look at significant prior probabilities across all diseases, we find that a single gene set is enriched across all of the diseases, KEGG_LYSOSOME. Lysosome activity is responsible for cell death, which is abnormal in all of the diseases we consider.
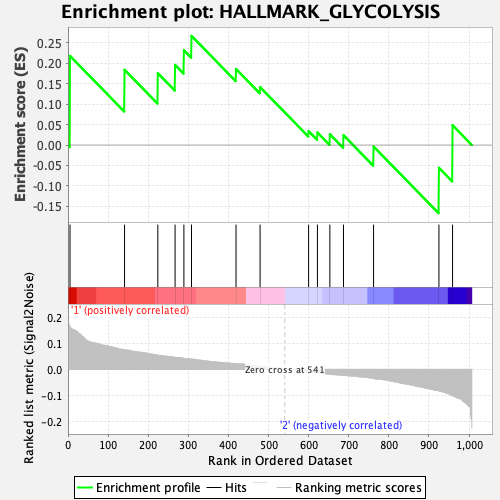
When we consider only regions with significant evidence of a shared causal variant, patterns begin to emerge that are unique to the disease type. When subsetting to a prior larger than 0.1, the gene set of the highest enrichment in the age-related diseases is HALLMARK_GLYCOLISIS. Literature shows glycolysis is abnormal in peripheral cells in Alzheimer's disease, Parkinson's disease, and Amyotrophic Lateral Sclerosis.
There are regions of significance between diseases that are individual to those diseases as well. When we isolate per-disease and subset by significant prior probability, the subset of genes indicates sources of genetic disease impact. For example, in osteoarthrosis we find that the HALLMARK_ESTROGEN_RESPONSE_EARLY and HALLMARK_ESTROGEN_RESPONSE_LATE are significantly enriched, indicating that hormone levels play a significant role in osteoarthrosis, which we know to be true from an empirical standpoint.
There is one region of overlap we see in the heatmap between hypertension and coronary atherosclerosis. This overlap is small but it indicates that there may be a shared disease mechanism. However, the region is so small that GSEA is unable to detect enriched pathways in the gene subset we extracted from that region using hierarchical clustering.
Principal Component Analysis of our colocalization yielded the right-skewed distribution expected in PCA. The first component accounted for around 60% of the variation, or in other words isolated one-third of our 6 diseases from the others, Alzheimer's and osteoarthrosis. This indicates that the majority of the variance can be explained by simple patterns, but a large amount is unaccounted for. Clusters generated using the PCA results seemed to isolate regions of significance unique to Alzheimer's and osteoarthrosis, but their corresponding gene subsets did not yield significantly enriched pathways when examined using GSEA.
Conclusion
Our results reaffirm that colocalization paired with GSEA is a beneficial strategy for understanding the genetic mechanisms of disease. Identifying lysosome activity as a significant factor against all diseases validates our pipeline since its results align with genetic and empirical facts. Moreover, identifying glycolysis as a mechanism in age-related disease in this manner is significant because the understanding of change in glycolysis activity was discovered through empirical observation. Seeing the same signal from a genetic standpoint in a case where the genetic basis was not the primary method of discovery indicates that other similar discoveries may be made in the future. Likely, purely genetic evidence discovered in this way can later be validated through empirical observation and form an invaluable foundation for disease prevention and treatment.